What is this for?
cREMaG is a database
interface allowing for detection of the over-representation of transcription
factor binding sites (TFBS)
in a queried set of
co-expressed genes. If the genes are co-expressed it is highly probable that
they are co-regulated.
Analysis of common
properties of their promoters could suggest the transcription factors
responsible for their co-regulation.
In this tutorial we will
not explain every function by detail. We will just pass through the whole analysis
process. The detailed help can be accessed by moving a mouse cursor onto
question marks as shown on the Figure 1.
![]()
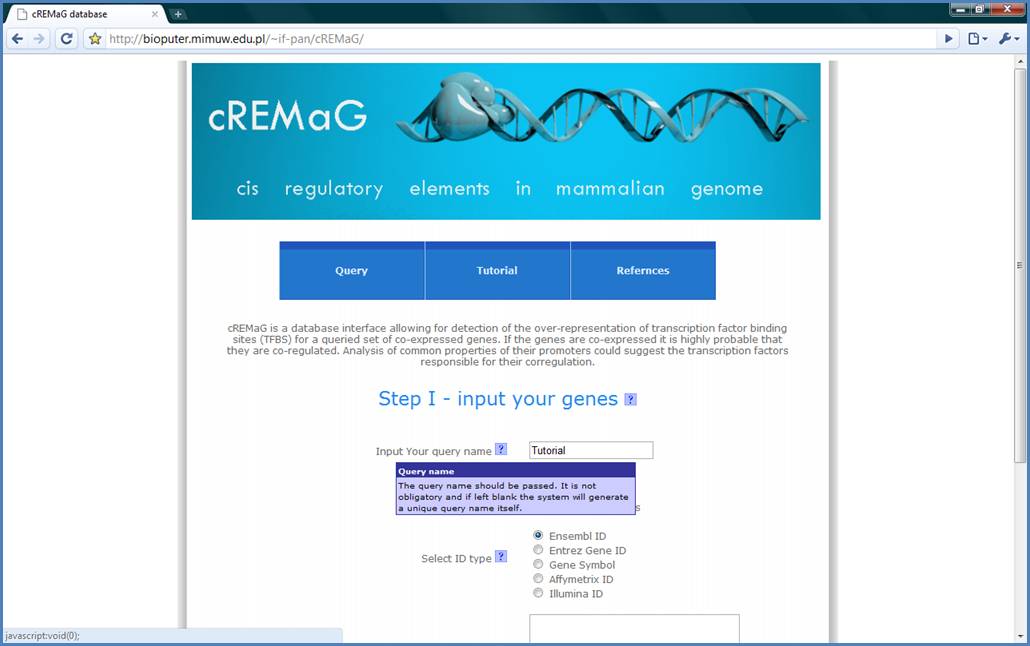
Figure 1.
First input your query name.
Please, fill the text field highlighted by the red box. We will use ‘Tutorial’
as a name of our query.
![]()
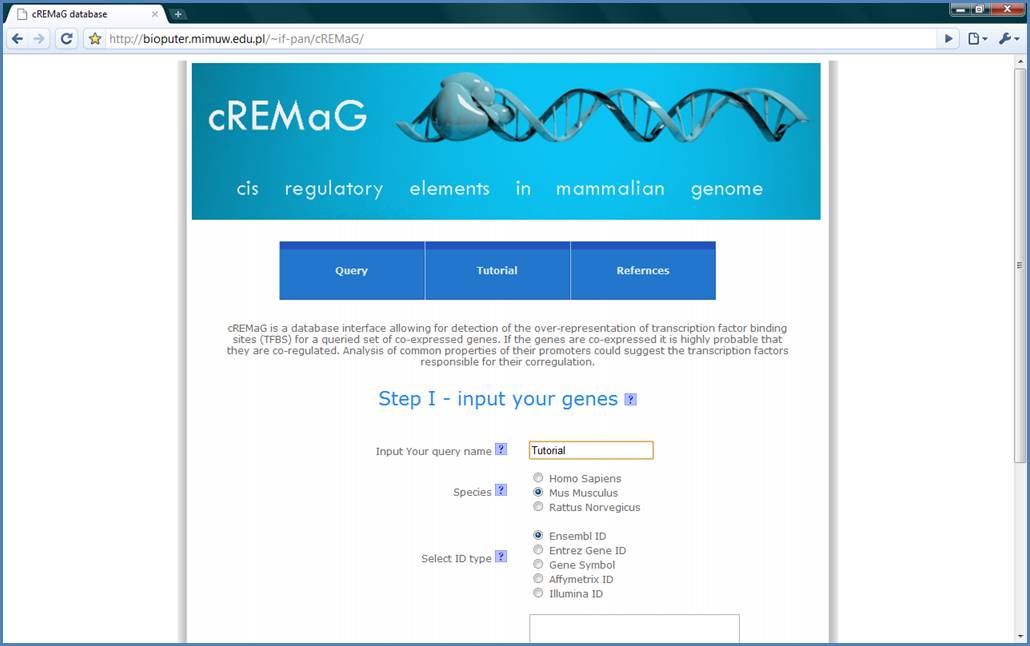
Figure 2.
Next, select ID species
and ID type. We will use Mus musculus and Ensembl ID for the tutorial example.
![]()
![]()
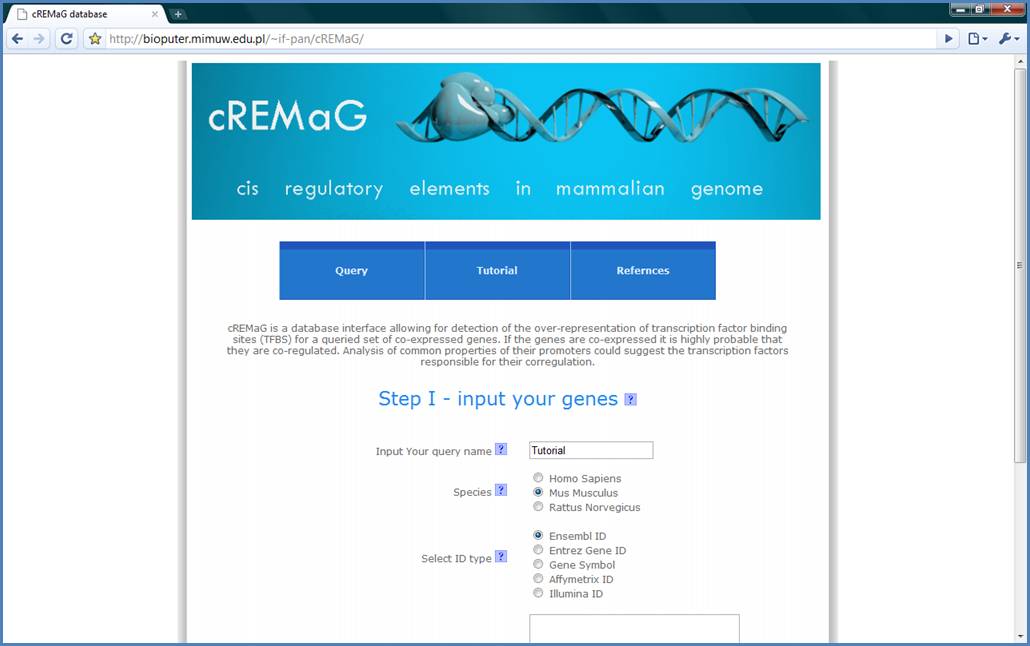
Figure 3.
Finally, we have to enter
our IDs. We have chosen Egr1, Egr2 and Egr4 genes for this tutorial as genes
controlled by SRF transcription factor (Ramanan N, 2005). cREMaG engine is
based on Ensembl IDs so it is recommended to use them. Ensembl IDs for the
chosen genes are ENSMUSG00000038418, ENSMUSG00000037868, ENSMUSG00000071341.
You can fill the text field by copying these IDs or by just pressing the
‘Example’ button. Finally we will focus only on the first transcription start
sites by selecting the Most Distal TSSs option. Now, You can press the ‘Next
Step’ button.
![]()
![]()
![]()
![]()
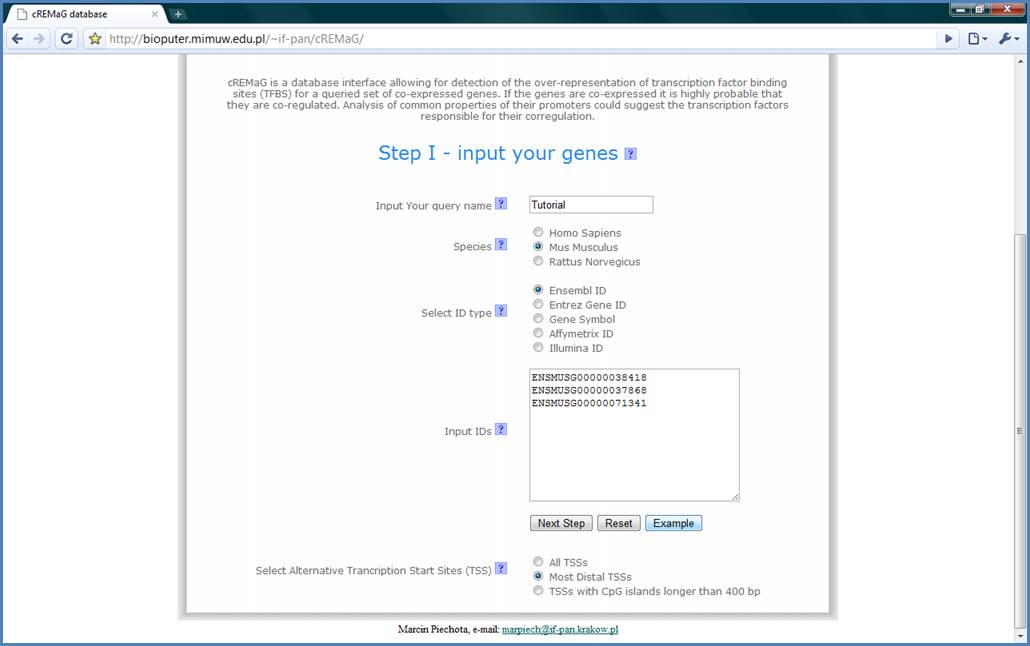
Figure 4.
In the Step II we have
information, which of the IDs were identified. For every identified gene we
have detailed information about its transcription start sites. For example for
Egr2 gene we have information what is the MGI Gene Symbol of this gene, what is
the Ensembl ID and what was the submitted ID. We have submitted Ensembl IDs so
the Ensembl Gene ID and Query ID are the same. Next, in a little bit brighter
box, there is information about the Ensembl ID of the homologous gene used for
phylogenetic footprinting. The phylogenetic footprinting is the procedure which
allows to obtain evolutionary conserved sequences. This is performed to reduce
noise in the analysis. Finally, we have brightest boxes with information about
the particular transcription start sites (TSSs): how many base pairs it is from
the first TSS (of course it is equal 0 if it is first TSS), how long are the
CpG islands around the TSS and what are the Ensembl IDs of particular transcript
forms. It is possible to select or deselect particular TSSs using checkboxes if
we want to add or remove them from analysis. Finally, please press the ‘Next
Step’ button. Please, notice that the second TSS (9bp) for the Egr2 is not
selected! We will come back to this in the Step III.
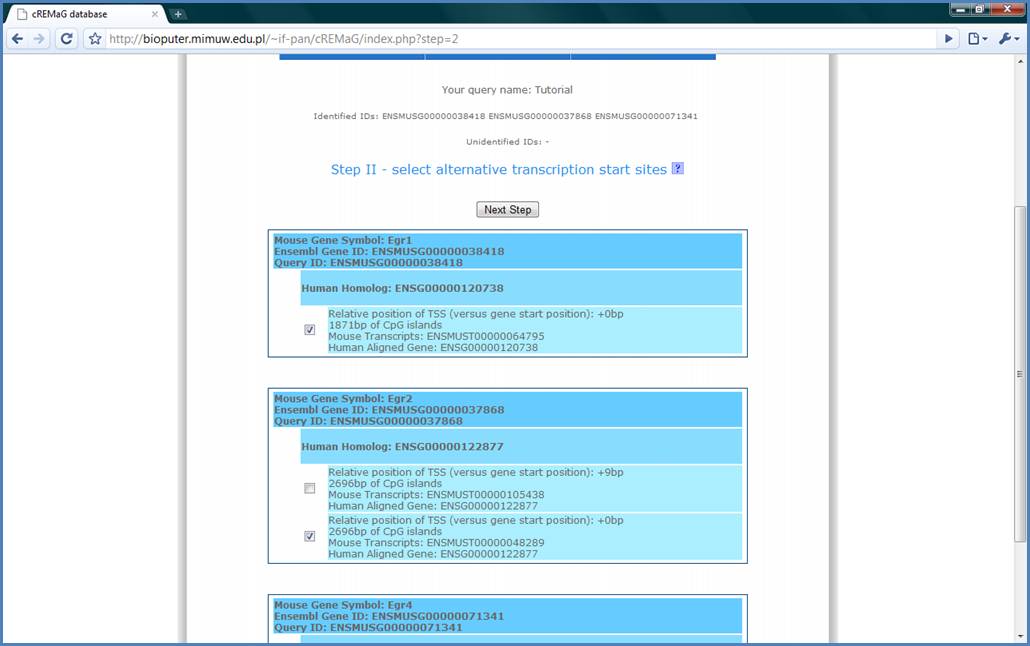
Figure 5.
In the Step III we have
multiple options, which are well described in popup windows after moving mouse
into them. We will pass to the next step using the default values.
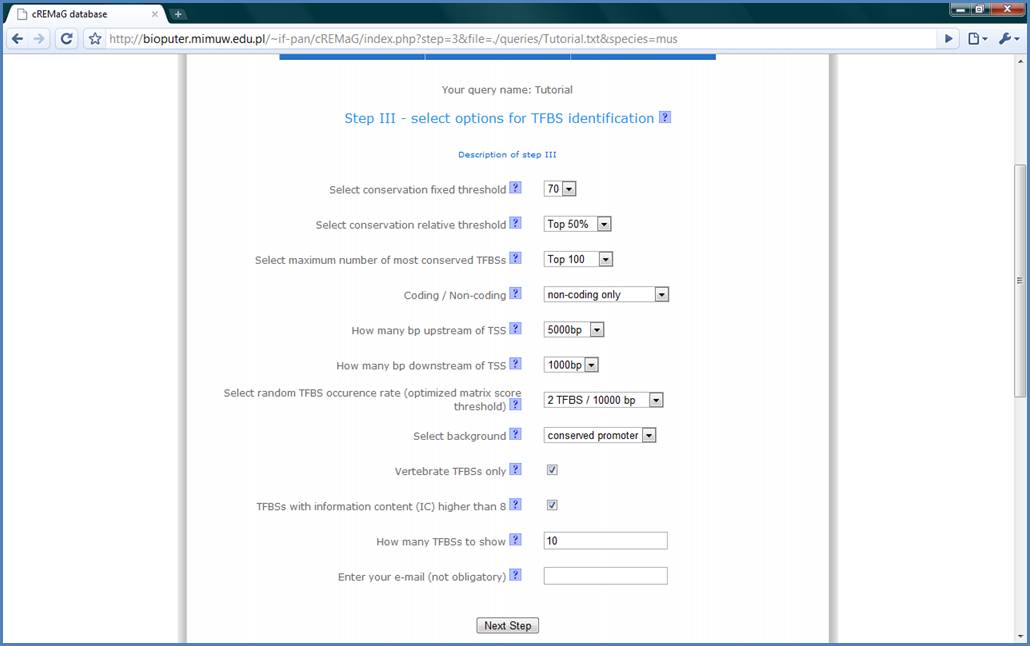
Figure 6.
In the Step III the information
about TSSs is repeated. The included (selected in the Step II) TSSs are marked
with yellow line, and excluded with red line. Please, press the ‘Next Step’
button after inspection.
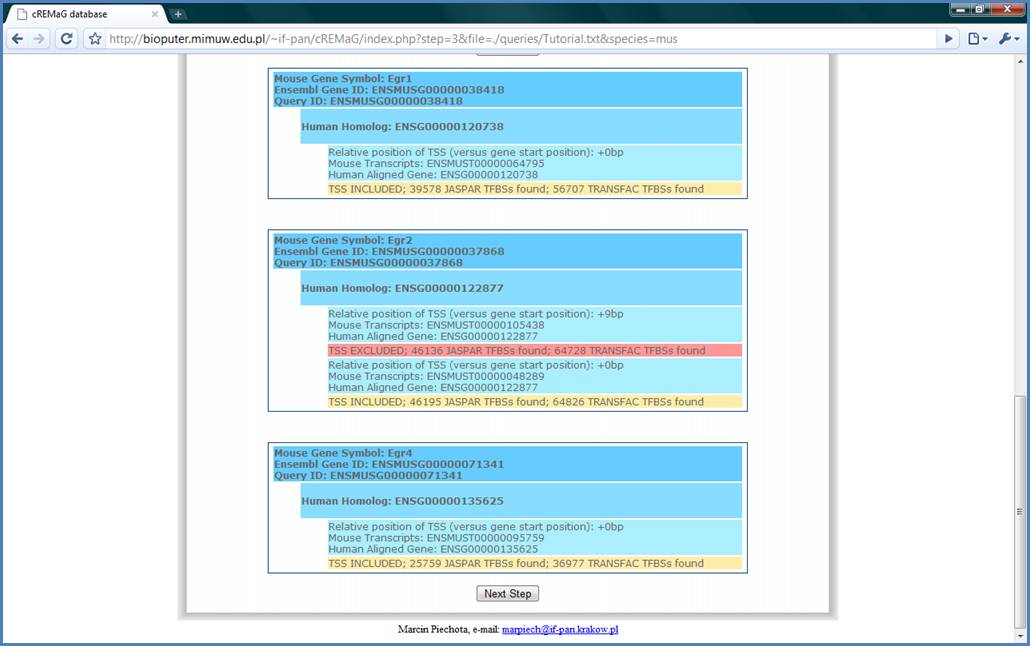
Figure 7.
Finally, we get the
results page. In the Query Info table, we have the selected options shown. In
Your genes table, the queried genes are shown and number of TFBSs on their
promoters which met the specified criteria (highlighted by the red box).

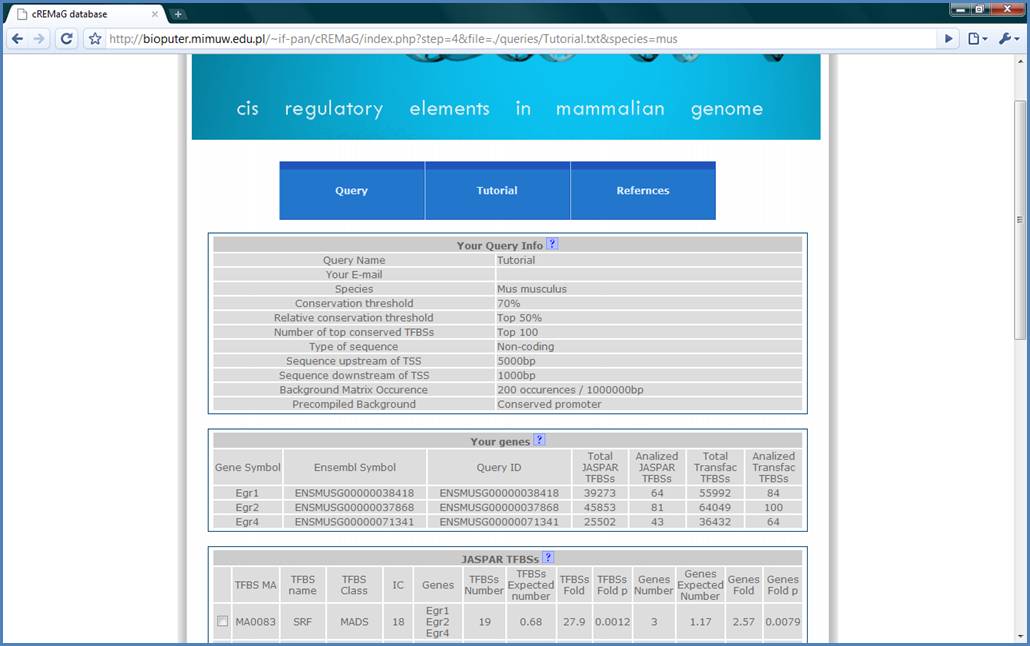
Figure 8.
In the JASPAR TFBSs Table
we have the most over-represented JASPAR matrices in our query gene set. The
most over represented binding site is SRF as expected (Ramanan N., 2005),
followed by ELK4, ELK1 and CREB1 which are also true positive results.

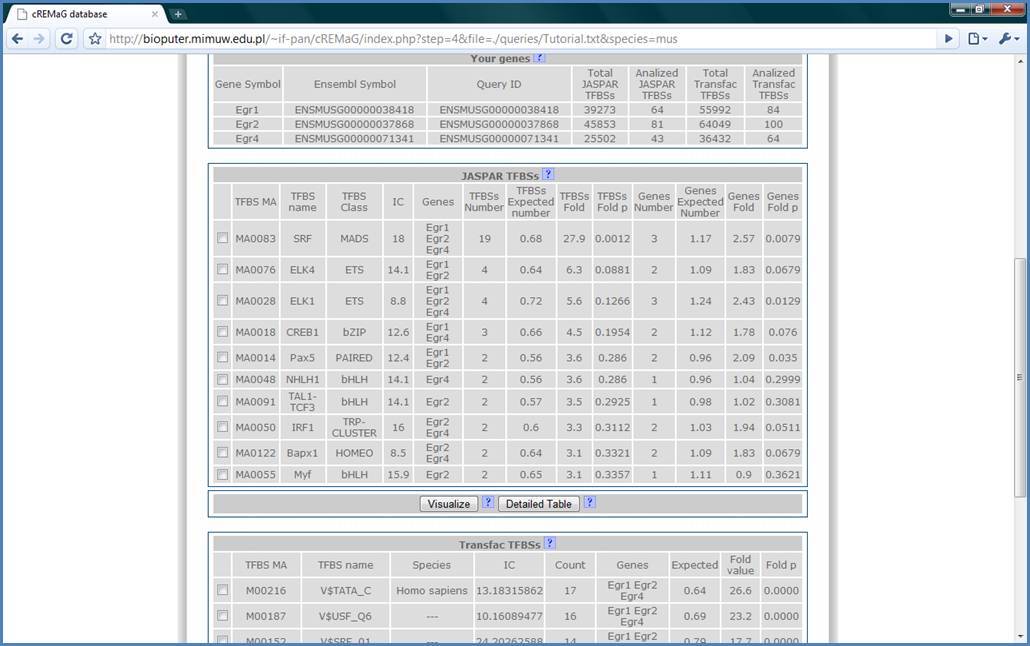
Figure 9.
The JASPAR TFBSs table
contains detailed information. ‘IC’ is the Information Content of matrix. The
higher the IC, the more specific matrix is. ‘Genes’ column contains symbols of
genes having the SRF binding site on its promoter. ‘TFBSs Number’ is the total
number of TFBSs found on the queried genes promoters and is compared to number
of TFBSs which would be expected by chance. Next, there is fold-difference and
p value of this difference. ‘Genes Number’ is the number of genes which have the
binding site on its promoter and is compared to genes expected number to get
fold-difference and its p value. High chance for getting true positive result
is when there is both TFBSs Fold p and Genes Fold p < 0.01.

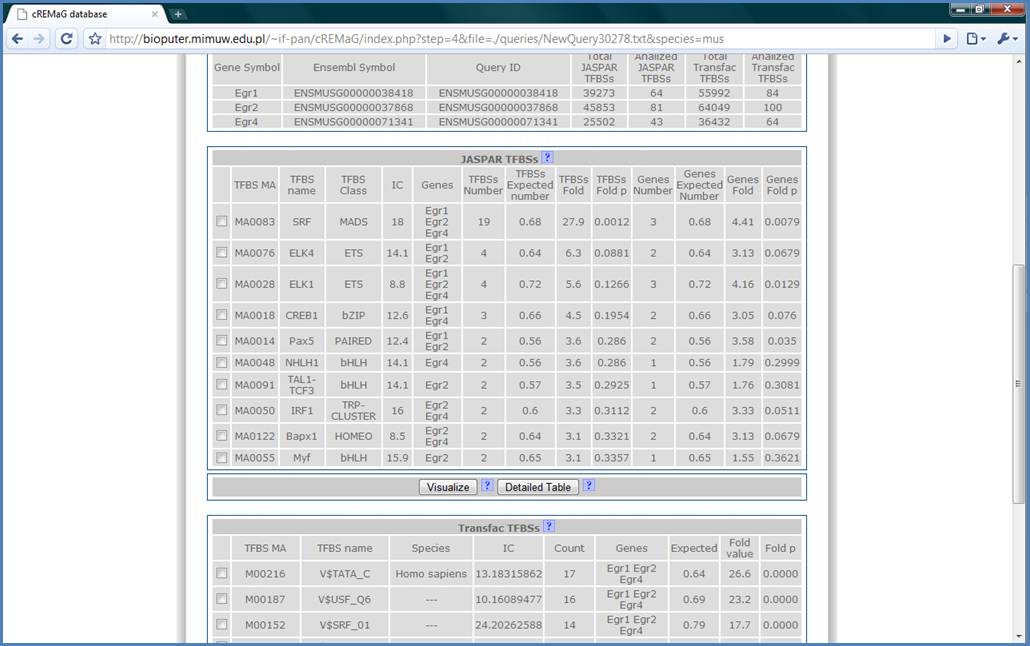
Figure 10.
You can also visualize
your results by selecting matrices to visualize by clicking checkboxes and
pressing the ‘Visualize’ button. We will select SRF and CREB1 for
visualization.
![]()
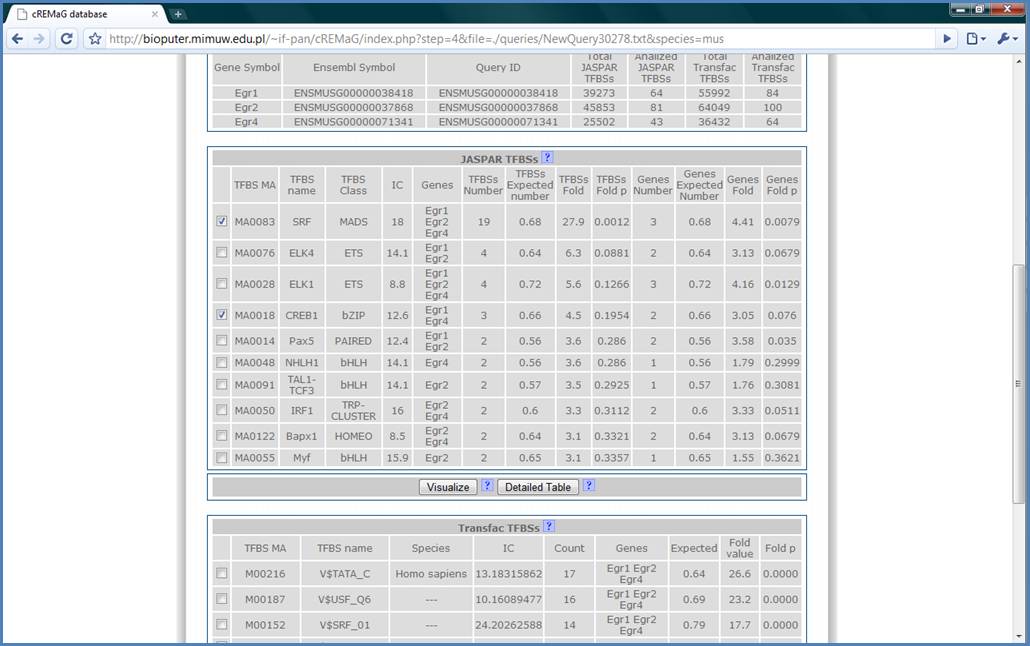
Figure 11.
The visualization screen
contains plenty of information which are not as well visible in text form. We
will not get into detail but it is clearly visible that binding sites for SRF (orange
ones) are situated on the highly conserved CpG islands on the core promoter.
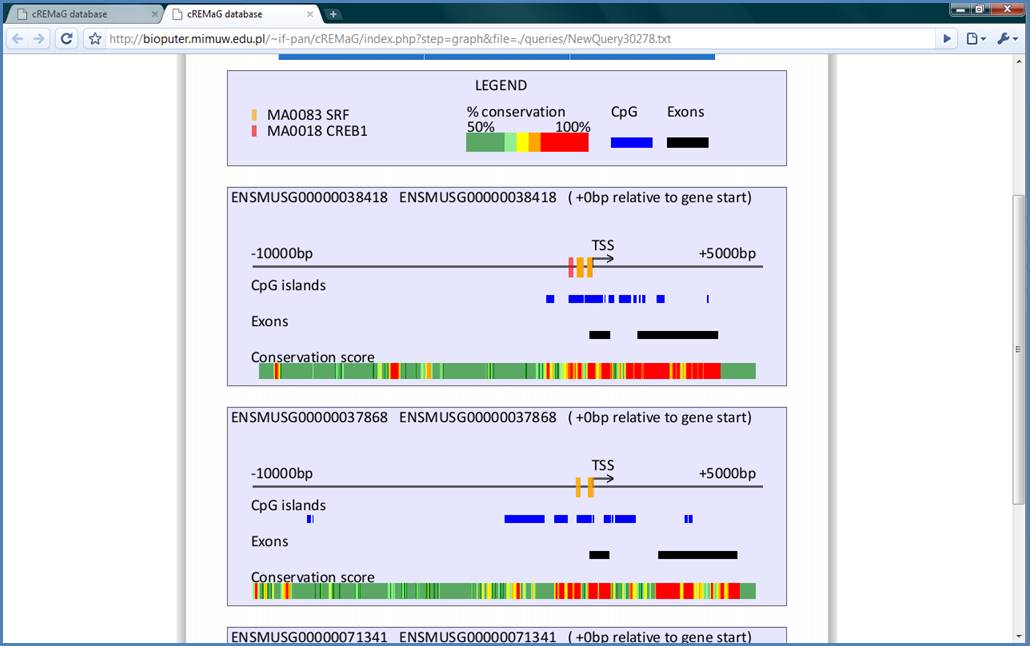
Figure 12.
You can also get the
detailed information about the binding sites by clicking the ‘Detailed Table’
button. We will use SRF, ELK1, ELK4 and CREB1 transcription factors to see the
detailed information, because there are working together as transcriptional
module.
![]()
Figure 13.
In the detailed view there
is position, score and conservation level of matrices. In such type of view it
is possible to identify regulatory modules as ELK1-SRF for example.
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
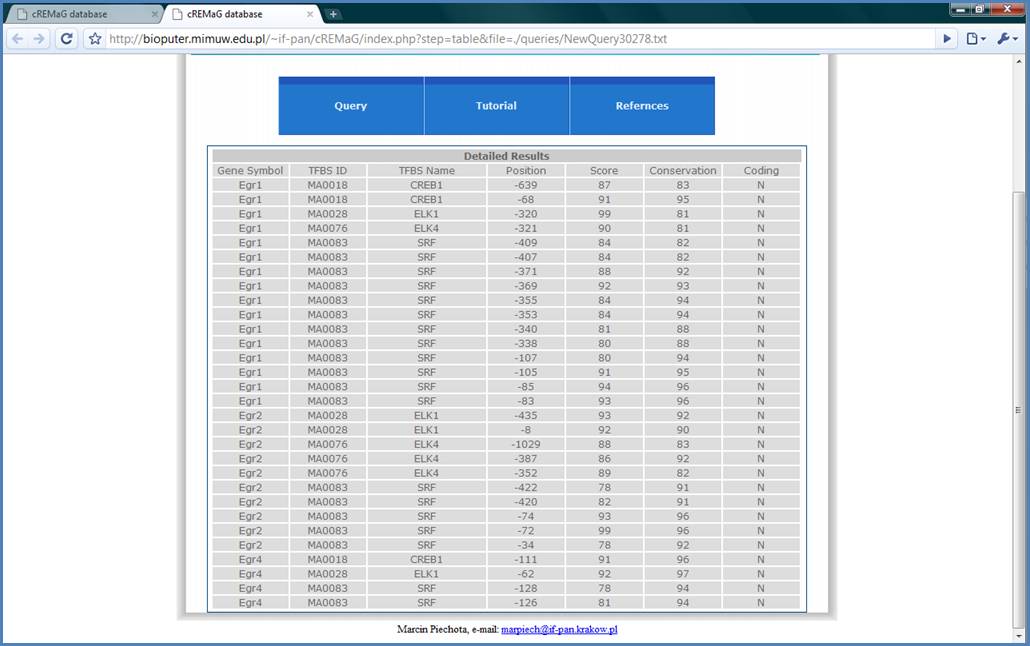
Figure 14.
Now You are ready to use
cREMaG. If You have any questions, please send e-mail to marpiech@if-pan.krakow.pl.